电子结构分析----文献翻译
一、前言
在网上翻到一篇讲电子结构分析的文献,内容比较基础,但是把概念讲的比较清晰,这里打算把全文翻译一下,原文链接:https://onlinelibrary.wiley.com/doi/10.1002/9783527671816.ch17
二、正文部分
电子结构分析
引言
你是否已经在你的模拟中使用过电子结构方法来计算你所选择的初始位置处原子的能量,并且当你移动原子到其它位置时,你也可以分析发生变化的电子结构。通过对电子结构的分析,你将更加深入地理解系统的化学键和稳定性。你也可以把你的把你的结果与很多实验结果对比,从而更好地理解甚至是解释实验结果。通常来讲,你会对扫描探针技术、光电发射、电子散射技术和磁共振等测试结果特别感兴趣。
为了计算结果合理,做电子结构分析时系统必须处于一个收敛态:很多现代计算软件包会使用很多方法来增加结构弛豫和分子动力学过程的综合性能,包括波函数和电荷密度的预测,如果仅靠分析这些不会给出可靠的结果。原子发生任何形式的移动之后,在分析之前,非常重要的一点是去运行一次单点计算得出原子的固定位置和高精度的电子基态。
对于模拟参数的理解也很重要。在一次DFT计算中,正确的收敛电荷密度尤为重要:例如如果你对能带结构感兴趣,最好首先在完整的k点集上计算一次收敛的电荷密度,然后固定住这个电荷密度,在它的基础上重新计算特殊点本征态和电荷密度。相比于结构弛豫,为了产生更好的电子结构,电子结构计算中使用更好的k点网格和更高的平面波截断能是有必要的。
1.能级和能带结构
在一个孤立系统中,例如,一个分子或者一个纳米晶体,能级大小与倒空间位置无关,因此只有一系列能级本身是我们所关心的。相反,在一个周期性系统中,根据凝聚态物理知识,你必须分析倒空间中不同位置处的能级。特别值得注意的是倒空间本身取决于原胞的大小和研究系统的性质。
实空间中一个高度定域化态,例如悬键或缺陷处的强束缚电子,在倒空间中将具有相应的平坦结构。物理上,这是讲得通的:实空间中的局域态说明和相邻晶胞之间相互作用很弱,因此波矢k的重叠很小。对于使用周期性边界条件来模拟非周期性系统,例如一个分子或者表面,这很重要:如果在某些物理上本应该是平的地方,能级随着倒空间位置发生剧烈变化,那么你应该检查你系统中所使用的原胞尺寸。
实空间中一个非定域化态将会产生显著的倒空间结构。最简单的例子是考虑一个自由电子金属,其能级下取决于倒格矢的平方,因为动能项包含k2因子。有一些金属已知是几乎近自由电子金属,尽管大体上是,但实际上它们更加复杂。注意半导体和绝缘体也有非定域态和可观的倒空间结构,并且仅需一点倒空间能带就能产生好的半导体模型。
实际上,我们已经接触到如何精确计算能带结构的问题。记住当你求解周期性系统的基态时,选取的离散k点集被用来近似计算能量的积分(这在能量最小迭代过程中是一个基础变量)。通过这样,能够产生一个良好收敛的基态电荷密度,你可以自信地用这个电荷密度很好地描述你的系统(前提你已经仔细地选取了相关参数)。这一步之后,电荷密度应该保持固定:如果你沿着特殊的k点集合计算能带结构,你不需要重复计算每个点的电荷密度(如果这样对基态电荷密度近似近似更差)。事实上,当你计算能级,你正在根据一个收敛的基态电荷密度,研究系统在倒空间中特定点的性质。使用哪种方法计算能带结构的问题应该是你计划阶段的一部分。一些能带结构是任何微扰计算(如GW)的必要部分,通常通过DFT执行,有时是Hartree-Fock方法。在执行计算之前,你必须确保输入的能带不包含任何严重的错误,包括输入文件的错误和使用的方法。对于DFT,通常会把带隙算的太窄,最需要检查的事情是本应该有带隙的体系,计算结果也应该有。
对于一个孤立系统,不需要考虑布里渊区采样点的问题,但是你仍然需要确保计算收敛到精确基态。这对于DFT和量子化学方法都是一样的,并且还需要仔细测试基组大小对能级的影响。
2.波函数和原子
在分析电子结构计算结果时,将电荷和其它性质投影到单个原子是非常有吸引力的。这也遵循标准的化学反映描述。然而,需要非常小心的是:没有明确的方法将空间区域分配给原子,而这些区域是计算电荷所必须的,这个问题的答案将根据使用的方法而改变。此外,对于给定的方法,随着方法所使用的参数变化,答案也会变化。这不是说你不需要尝试去把电荷投影到原子:只是说你需要清楚自己在做什么和为什么做,并且对结果持谨慎态度。
为什么有这样的问题?最简单的出发点是询问关于怎样定义和每个原子相关的空间体积。最典型的回答是去使用一个球形,这很有道理,因为孤立原子具有球对称性。然而,从水果可以看出来,当你堆叠这些球时,它们周边存在间隙,你不能用球填满空间。因此,当使用球来填充空间时,我们将会损失间隙处的电荷密度,或者我们必须让球重叠并且将某些相同的电荷密度分配给不同的原子。
目前有一些方案来区分原子间的重叠区域,通过给定不同的权重,虽然这些都带有一定的经验注意。这其中两个最著名的方法是Hirshfeld方案和Becke方案。Hirshfeld方案提出了一种“promolecule”的电荷密度,它是指孤立原子基态下的电荷密度,然后对根据空间中每个点对电荷的贡献进行加权(因此我们可以得到原子A在r处的电荷比重为:wA®=ρA®/∑BρB®)。Becke方案使用积分方案一部分作为权重,这在很多量子化学代码中已经是标准。它根据Voronoi多面体的推广定义了原子边界。它的主要优点是,它通常已经用于积分过程中,并且可以用于计算原子电荷,而无需额外的计算成本。
重叠球体的另一种选择是使用与球体不同形状,它可以完全填充空间。有很多方案来做这件事情,尽管同样的,由于划分的方法本身并不明确,因此不同的方案将会产生不同的结果。Bader atoms-in-molecules 方法根据电荷密度的拓扑结构确定原子间电荷过渡的位置。这需要完整的电子密度,如果使用赝势,必须要重构。这一方法主要是搜索沿着键的方向电荷密度最小,垂直键的方向最大的点的集合。这在计算上花费较大,并且有时会给出很难懂的结果,尽管非常有效并且被广泛应用。最常见的方法是使用Voronoi多面体方法,它们是用原子对之间的向量平分的平面来定义的。平面包围的空间指定给位于空间中心的原子。这种方法不像许多其他方法那样考虑不同的原子大小,尽管目前已经有引入原子大小的扩展。
可用于定域基组的最后一种方法是通过密度矩阵P,在原子轨道基组中,来分配电荷密度,尽管这种方法也需要特别注意处理重叠矩阵S。被广泛应用的Mulliken方法使用乘积元素PS,一个原子给定的电荷定义为:PiiSii+1/2∑j(PijSij+PjiSji),在原子上对基函数进行隐式求和。Mulliken方法的关键问题是其它元素的加和因子1/2:这是任意的。Lowdin方法转而使用正交基矢,并且使用生成的电荷密度矩阵S1/2PS1/2的自身元素计算。Mulliken方法对基组的依赖度很大,对于较大原子基组结果将变得不可靠并且不收敛。
如下表给出了一些使用不同方法计算得到的结果。很容易看到,不同的方法计算结果差距很大,并且受使用的基组影响很大,Mulliken方法改变符号随着随着基组增加,Bader电荷随着基组变化逐渐收敛,Hirshfeld方法和Voronoi方法几乎与基组选择无关。这一简单例子能够清晰的说明给原子分配电荷本质上是有风险的。
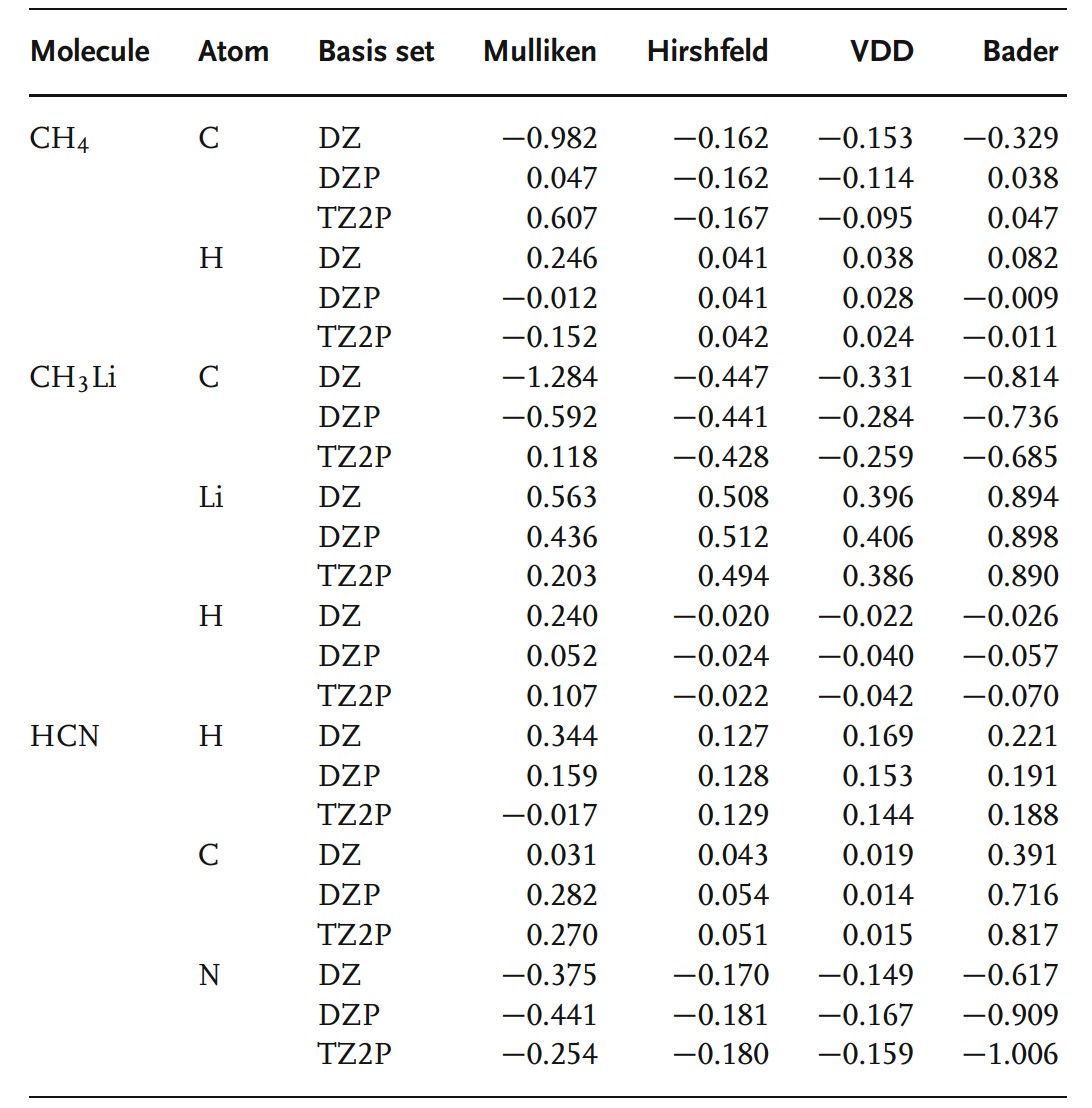
因此我们应该选择什么方法呢?我们不能给出具体的建议,因为这些方法都有弱点,在使用Mulliken方法的时候我们必须慎之又慎,因为它非常不可靠。如果你不使用原子中心化局域基组,你必须使用更多的方法来分割空间。无论使用何种方法,我们建议你做一下重要的检查:测试你的假设的效果;不同的计算结果自洽;不要过度解读。
3.定域化函数
将一组扩展轨道转化为定域函数是电子结构方法中的一项重要技术。对于扩展系统,生成的函数称为Wannier函数,而对于孤立系统,一般称为局域分子轨道。然而,仅仅规定有一个幺正变换是不够的:必须提出一些更进一步的标准。对于扩展系统,一个非常成功的方案被提出来去生成最大定域Wannier函数,通过减少数目:Ω=Σi<Φi|r2|Φi>-<Φi|r|Φi>2,其中沿着模拟晶胞中的能带求和。对于分子轨道适用的Foster-Boys定域标准可以证明形式上是一样的,尽管求和是沿着分子中的轨道。还有其它的标准定义,例如最大库伦相互作用或者轨道电荷重叠。都是后处理程序,需要从自洽的收敛电荷密度开始。
一旦创建了定域化轨道,它们可以被用作分析系统。最显而易见的和广泛的应用是分析化学键。一些程序中保留了σ键和pi键的成键特征,通过能量区分,而另外一些混合它们。然而,与原子间键相关的定域轨道可以被观察到并被用于理解分子和晶体中相互作用。在周期系统中,它们还可用于计算材料在外场作用下的极化,这是用非定域化函数无法实现的。
定域轨道也可以被用作计算电子输运的基函数,尽管一些方法简单地一开始就使用原子中心基函数,计算输运是一个广泛的领域,对于不同问题有很多不同的方法。在最简单的情况下,采用散射方法,利用非平衡态格林函数,可以扩展为自洽、存在电子传输的情况下来计算电荷密度。目前有很多程序能够将DFT计算结果与非平衡态格林函数方法提供接口计算输运性质。这种方法可以研究电子跃迁,而不是相干电子传输,但这涉及电子的定域化和计算Born–Oppenheimer表面之间的跃迁。
定域轨道也可以被用作计算相关性。在量子化学中,一些减少关联计算比例的方法涉及定域轨道,并且将这些方法应用于几十个原子已经成为可能。在扩展系统中,Wannier函数被用作哈密顿量模型用于强关联系统的计算。
4.态密度,投影态密度
态密度(DOS)是一种测量在特定能量下,或更准确地说在特定能量窗口中有多少态,即电子结构计算中的本征态。DOS可以与实验技术关联,例如STM,也可以用于分析系统的电子结构。将DOS分解到原子或原子团也是有用的,尽管和向原子分配电荷一样,也有一样的困难,下面将详细讨论。形式上讲,我们可以写作:n(E)=Σiδ(E-εi)
在DFT计算之后,我们可以通过简单地将窗口划分为一组存储单元并计算每个存储单元中的状态数来评估能量窗口中的DOS。DFT处理有限数量的原子,因此最终DOS将会离散。对于这样的一个有限系统,DFT计算得到的DOS将只会简单地是一组竖线,与实验光谱几乎没有什么相似之处(但是对于0K下地分子系统是相似的)。
通常的做法是通过离散的本征值和用户指定宽度的高斯函数来加宽它们生成DOS图。这在物理上讲的通:热涨落将会拓宽系统的能级。然而,这意味着生成的DOS的整体外观可以取决于用户的输入。下图中说明了加宽效应对DFT计算的苯的DOS的影响。这里,我们可以看到非常小的宽化,非常窄的峰,但是保留了所有的系统能级的细节。随着宽度增加,可能在视觉上看起来更好,但是峰值开始合并。随着更大的展宽(smearing),两个峰可能会组合成一个。可以注意到垂直比例怎样随着展宽变化的:使用的约束条件是DOS积分得到总的态度米,增加展宽之后必然会降低峰值高度,因此在选择参数时需要谨慎。
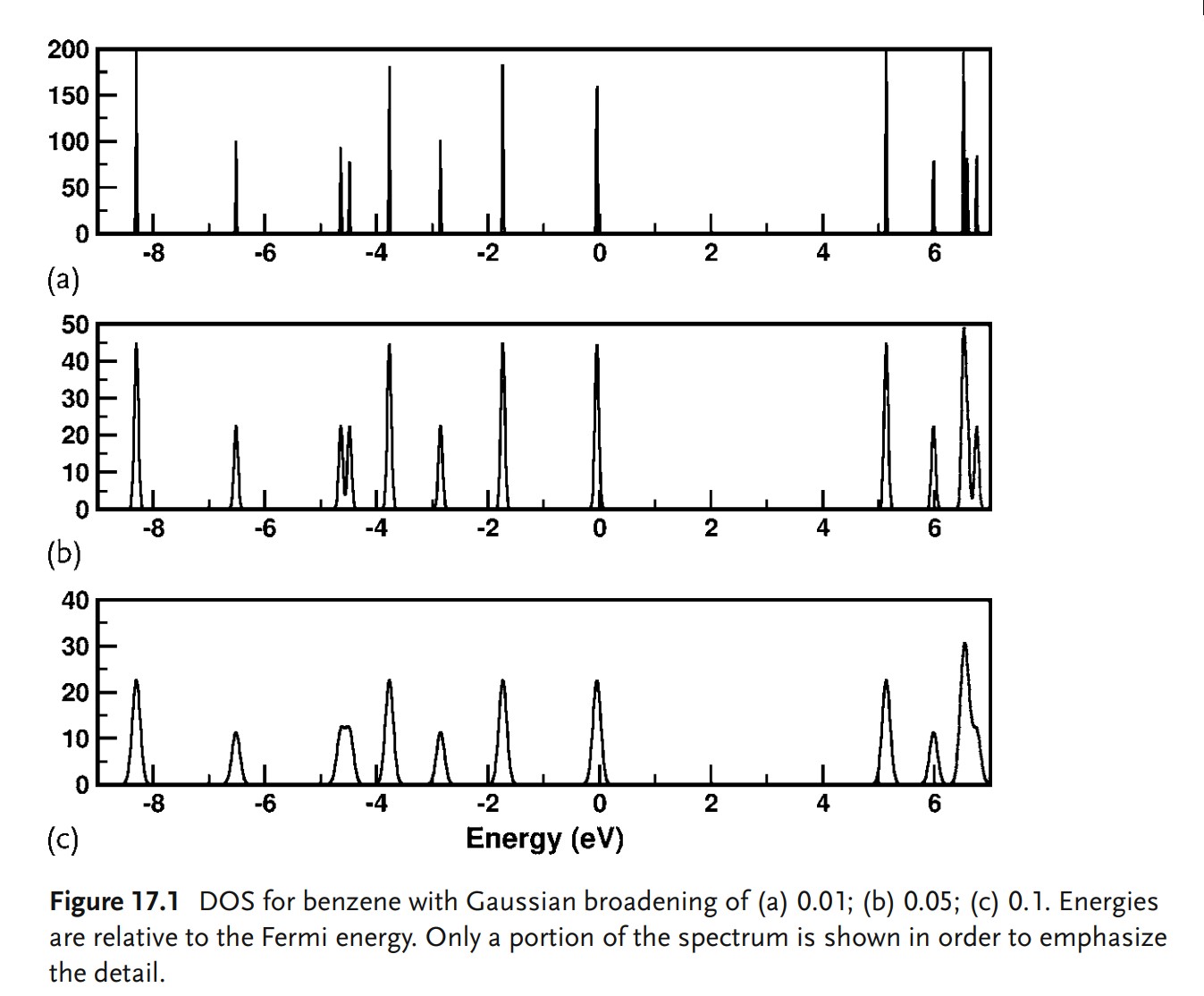
当考虑具有局域特征的系统的物理或化学性质时,例如表面,缺陷,反应中心,整个系统的DOS常常不是很有用。在这种情况下,很自然地会想,与特定原子或原子组相关联的DOS可能是什么。将DOS投影到特定原子上的问题与前面讨论的将电荷分配给特定原子的问题完全相同:在DFT中,波函数是非定域的,没有明确的方法将波函数分配给特定的原子。最常用的方法是在原子周围定义一个球体,并将DOS投影到该球体上;重要的是要认识到,产生的DOS将取决于球体半径。这并不意味着你不应该这样做:简单地说,就像电荷一样,你需要测试你的假设的效果并保持自洽。