VASP中POTCAR使用指南
POTCAR是VASP计算的四大输入文件之一(INCAR、POSCAR、POTCAR、KPOINTS),官方一般会随着新版本的VASP软件包发布的同时提供相应的各个元素的POTCAR库,一般计算时直接根据元素类型组合一下使用就行了,使用并没有什么难度。但是,觉得有必要写一下这篇文章主要是基于以下几点考虑的:
- 使用POTCAR的好坏对于计算结果的好坏影响很大,远比INCAR,KPOINTS中参数设置的好坏影响大
- 对于计算不同的体系和不同的性质,选取何种类型的POTCAR也有讲究,需要仔细斟酌
- 基于GGA近似后来发展出来的meta-GGA和HSE方法,计算某些性质时精度更高,但同时更加耗时,平衡好计算时间和计算精度是必须要考虑的问题(如果你的超算资源非常充足当我没说)
Jacob的天梯说明了交换关联能不同近似方法的精度高低,下图形象说明了这一问题: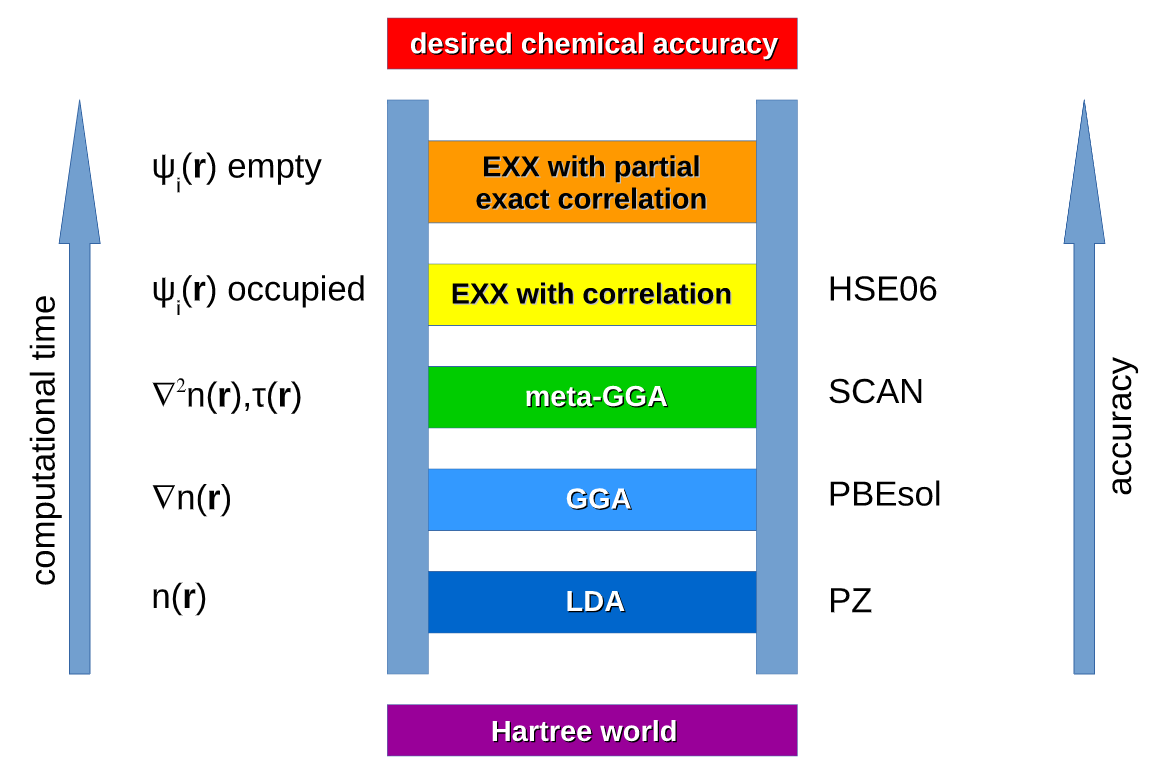
1.POTCAR简介
这部分内容主要是来自VAPS官方手册。
POTCAR文件包含计算中每种类型原子的赝势,如果原子类型数量大于1,需要将它们连接起来,Linux系统中使用如下命令:
1 | cat ~/pot/Al/POTCAR ~/pot/C/POTCAR ~/pot/H/POTCAR > POTCAR |
来连接POTCAR文件,第一个文件对应POSCAR第一个出现的原子类型。
注意:如果POTCAR文件的连接顺序与POSCAR中不一致,VASP会按照赝势文件中给出的顺序来识别不同元素,因此计算结果是错误的
对于3.2以上版本的VASP,POTCAR文件包含这些元素的信息,例如:它们的质量POMASS,它们的价电子数ZVAL,创建赝势时相应参考构型的能量等。如果在INCAR文件中又重新指定了质量和价电子数,则VASP会根据POTCAR文件中的参数检查它们,并且输出相应的错误信息。POTCAR文件中也包含默认的截断能(ENMAX和ENMIN)。因此在INCAR文件中指定ENCUT是非必须的。在INCAR文件中ENCUT的设定值会覆盖POTCAR中的默认值。对于包含多个元素类型的POTCAR文件,将会使用最大ENCUT值用于计算。
重要:POTCAR文件旨在以只读的方式提供给用户使用,VASP没有需要修改POTCAR文件的标准用法,尤其是不要修改POTCAR文件中的LEXCH标签:它是VASP使用INCAR中指定的函数重新计算PAW球内交换-相关能量的原子校正能量。为了使VASP正常工作,POTCAR文件中的原始LEXCH标签值不允许被修改。
2.提供的PAW赝势
这部分内容主要来自这里。
从VASP官网可以获得周期表中所有元素的投影缀加波(PAW)势文件,这些是PAW方法的赝势,存储在POTCAR文件中,G.Kresse按照文章中的方法生成了分布式PAW势文件,而Peter Bloch首先提出了PAW方法。因此如果你使用任何提供的PAW势文件,你应该引用这两篇文献。
除了周期表中第一行元素,所有的PAW势都被设计成在大约250eV截断能左右能够可靠和精确地运行。这是使计算成本降低的关键层面,默认的截断能是通过POTCAR文件中的ENMAX标签设置的,PAW势相较于超软赝势(US-PP)更加精确,有两个原因:第一,相较于US-PP,PAW势使用的截止半径(芯电子半径)更小,第二,PAW势重构了芯电子区域的所有节点的精确波函数。由于PAW势的芯半径更小,因此所需要的基组和截断能更大,如果这么高的精度没有必要,原则上旧的US-PP势是可以使用,但是并不鼓励,这是因为对于C、N和O,截断能并没有明显减小,同时,基组通常增加得很少,因此使用PAW势计算包含这些元素的化合物不会比使用US-PP更昂贵。
对于一些元素,存在多个版本的PAW势文件,标准赝势文件名称没有扩展,对于含有其它不同扩展的版本,相应的含义如下:
- _h扩展:表明当前势文件相较于标准势文件更硬,因此需要更大的截断能
- _s扩展:表明当前势文件相较于标准势文件更软
- _pv扩展:半芯态p电子被当作价电子处理
- _sv扩展:半芯态s电子和p电子(如果有)被当作价电子处理
- _d扩展:半芯态d电子被当作价电子处理
每个PAW势中采用的价电子构型可以通过POTCAR中的ZVAL标签和Atomic configuration下的表格推测出来。这一表格首先列出来了芯电子态,然后是价电子态。因此,这些占据态的最后一行加起来正好是ZVAL,组成了价电子构型。注意,这可能与真空中的基态构型不同,并且这些行不是按能量排序的,例如,尽管Gd的强定域化、半芯态3f电子能量比其它价态电子能量更高,但是仍然被当作芯电子处理。
接下来,我们将展示可以获得的PAW势。所有的这些分散的势文件都已经通过标准的DFT-"benchmark"运行测试过。具体可以参阅公布的tar文件中的data_base文件,我们强烈建议使用可在VASP官网上下载的5.4版的POTCAR文件,目前分散的5.4版本的POTCAR文件都有一个独一无二的SHA哈希值,5.2版本的POTCAR文件也很流行并且已被Materials Project使用。5.4版本和5.2版本的之间的差别很小,并且只限于少量的元素。目前可以从官网获得的5.2版本的每个POTCAR文件同样有一个独一无二的哈希值,并且它们在标题的文本部分被编辑过,但这与VASP计算无关。
3.DFT计算中推荐使用的势(PBE计算)
a.DFT计算推荐的势文件(PAW)
如果化合物中包含短键的二聚体(O2, CO, N2, F2, P2, S2, Cl2),我们建议使用_h势文件,即C_h, O_h, N_h, F_h, P_h, S_h, Cl_h。
在5.4版本,W_sv代替了W_pv,并且At_d的POTCAR文件不再提供,因为这一势会导致计算的晶格参数出现很大的错误。
类氢提供了价电子数从0.25到1.75的势文件,注意POTCAR文件限制了价电子数的位数(有些是2位,大部分3位),也就是说,用三个H.33势不会产生0.99或是1.0个价电子,这可能会产生无法预料到电子-空穴态,解决办法是稍微调整INCAR文件中的NELECT标签。
b.GW/RPA计算推荐的势文件
GW势计算结果与上述DFT计算结果几乎相同,因此使用GW势计算并不会使得上面DFT的计算结果恶化,事实上,通过对比我们已经证明在全电子计算中,GW势相比于上面DFT计算更有优越性。它们对于激发态特性、GW计算、随机相位近似计算(RPA)、以及任何显式相关的波函数计算(MP2, 耦合簇)通常更有优势。
通常,GW势在远高于费米能级的高能量区间能产生更好的散射特性,尤其是比真空能级高10-20Ry的能量区间。
重要提醒:如果是化合物中包含短键的二聚体(O2, CO, N2, F2, P2, S2, Cl2),我们建议使用_h势文件。
C_GW_new、N_GW_new、O_GW_new和F_GW_new势文件使用f-赝势作为局部势并且拥有d-投影,相反,C_GW、N_GW、O_GW和F_GW势文件使用d-赝势作为局部势,不包含d-投影,在使用ENMAX作为截断能的情况下,使用C_GW、N_GW、O_GW和G_GW势通常收敛更快,相较于旧的势文件,新的势文件是否在精确性上更有优势还没有完全搞清楚。理论上,它们在相关波函数的计算上应该更精确,然而,实践中发现,提升似乎不明显,通常并不能证明更大的计算负载是合理的。
c.关于PAW势的进一步建议
第一行元素
对于第一行元素,存在三个PAW版本的势文件,对于大多数目标,标准版本的PAW势是合适的。在截断能范围为325-400eV时,它们能够得到可靠的结果,要预测精确的振动性质需要截断能范围在370-400eV。结合结构和能量差异在325eV处就可以很好地重现。与更精确的DFT计算相比,第一行二聚体(N2,CO,O2)预测的典型键长误差约为1%。_h硬赝势给出的结果与目前可用的最佳DFT计算软件(FLAPW,或具有非常大基组的高斯)基本相同。软赝势在截断能250-280eV区间使用,对于大部分氧化物它们能得到可靠的描述,例如VxOy、TiO2、CeO2,但是在描述沸石的某些结构细节上失败了,如晶胞参数,体积。
对于Hartree-Fock(HF)和杂化泛函计算,我们严格建议使用标准、标准GW或硬赝势计算,例如,在过渡金属氧化物计算中,O_s赝势可能造成不可接受的巨大误差。一般来说,软势不太可能从一个交换相关函数转移到另一个函数,并且在需要计算精确交换时经常失败。
碱和碱土金属(简单金属)
对于锂(和铍),提供了标准势和把1s壳看作价电子的势(Li_sv, Be_sv),在应用中应该使用_sv势因为它们比标准势的可移植性更好。
对于其它碱和碱土元素,半芯s和p态也应该被当作价态处理。对于更轻的元素(Na-Ca),将2p态或3p态分别当作价态处理通常还不够。对于Rb-Sr,4s、4p、5s和5p态都必须被当作价态处理(_sv)。
与普遍看法不同的是,这些元素在与强电负性元素如F结合时,非常难赝化,误差可能比平时大。
目前提供的赝势非常精确,应该提供非常可靠的描述。对于X_pv赝势,半芯态p电子被看作价电子,例如:Na和Mg的2p电子,K和Ca的3p电子等。对于X_sv势,半芯态s电子被看作价电子,如Li和Be的1s电子,Na的2s电子。
d区元素
与碱和碱土元素一样:半芯态p和半芯态s同样也应该被当作价态。在大部分情况下,然而,即使冻住半芯态,也能得到可靠的结果。根据经验,当p态的本征能量ε高于3Ry时,它应该被看作价态。
对于早期的过渡金属,需要使用X_pv势,但是后期的过渡金属,可以冻住半芯态p,尤其是贵金属。何时从X_pv势切换到X势取决于所需的精度和3d元素所处的行,即使是Ti、V和Cr的标准势能给出合理的结果,也应该极其小心地使用。4d元素的问题最大,我建议直到Tc都一直使用X_pv势。对于5p元素,5p态定域化非常强(低于3Ry),因为4f层被填充了,从Hf开始可以使用标准势,但是我们建议先测试一下。对于有些元素,X_sv势也是可以使用的(如Nb_sv, Mo_sv, Hf_sv)。这些赝势的截断能通常与_pv赝势相似。对于HF-型和杂化泛函计算,我们强烈建议尽可能使用_sv或_pv赝势。
p区元素(包含第一行)
对于Ga, Ge, In, Sn, Tl-At,d态应该被看作价态(_d势)。
对于这些元素,将d态看作芯态的势文件也可以使用,但要特别小心。
f区元素
由于本身相互作用的误差,目前密度泛函方法还不能很好地处理f电子。尤其是,不能正确描述部分填充的f态,例如所有的f态都固定在费米能级处,导致Pr—Eu和Tb-Yb之间存在较大的过度束缚,在f电子处于四分之一或四分之三填充时误差最大,例如,Gd得到了很好的处理,因为7个电子占据了大部分的f壳层。这些误差是DFT本身的原因,而不是VASP的。特别有问题的是描述在每个阶段开始观察到的巡游行为到阶段结束时的定域态。对于4f元素,这种转变已经发生在La和Ce中,而对于5f元素,转变发生在Pu和Am中。解决当前DFT无法描述4f定域化电子的常规方法是将4f电子置于芯电子中,这些势是可用的,但是它们在描述f轨道的磁性上会失败。此外,将f态当作价态处理的PAW势也是可以使用的,但是当f电子是定域化时,它们在描述电子性质上会失败。在这种情况下,可能需要更加小心地处理电子的相关效应,例如,通过使用杂化泛函或者引入原位库伦相互作用(+U)
对于一些元素,也提供了更软版本的势文件。半芯态p经常被看作价电子,而半芯态s只有在标准势文件中才被当作价态。对于大部分应用(氧化物,硫化物),应该使用标准版的因为更软版本势文件可能在靠近费米能级处产生s幽灵态(例如Ce_s)。对于计算金属间的化合物,更软的赝势版本可以达到足够的精度。
此外,还为Ce到Lu提供了特殊的GGA势,其中部分f电子被冻在芯电子中,这是一种处理f电子定域化的尝试。芯中的电子数等于总的价电子数减去形式化合价。例如:根据元素周期表,Sm总共有8个价电子,即6个f电子和2个s电子。在大部分计算中,Sm采用3价,因此在产生赝势文件时5个f电子被放入芯中,对应的赝势文件可以在Sm_3目录中找到,化合价n通过_n来标记,这里n可以是2或3。例如,对于Ce_3,是三价Ce对应的赝势文件(对于四价Ce,应该使用标准赝势文件)。